





 Corosolic acid induces apoptosis through mitochondrial pathway and caspases activation in human cervix adenocarcinoma HeLa cells
Corosolic acid induces apoptosis through mitochondrial pathway and caspases activation in human cervix adenocarcinoma HeLa cells | Corosolic acid induces apoptosis through mitochondrial pathway and caspases activation in human cervix adenocarcinoma HeLa cells |
| ����ʱ��:2012-10-11 ��Ϣ��Դ:admin ������:admin �������:6599 |
Corosolic acid induces apoptosis through mitochondrial pathway and caspases activation in human cervix adenocarcinoma HeLa cells abstract We investigated the response of human cervix adenocarcinoma HeLa cells to Corosolic acid (CRA) treatment. Our results showed that CRA significantly inhibited cell viability in both a dose- and a time-dependent manner. CRA treatment induced S cell-cycle arrest and caused apoptotic death in HeLa cells. We found that CRA increased in Bax/Bcl-2 ratios by up-regulating Bax expression, disrupted mitochondrial membrane potential and triggered the release of cytochrome c from mitochondria into the cytoplasm. Moreover, CRA treatment triggered the activation of caspase-8, -9 and -3 in HeLa cells. All these results indicate that CRA-induced apoptosis is associated with the activation of caspases via a mitochondrial pathway. Taken together, we believe that CRA could have strong potentials for clinical application in treating human cervix adenocarcinoma and improving cancer chemotherapy. Keywords: Corosolic acid Cervix adenocarcinoma Apoptosis Caspase Mitochondria 1. Introduction Apoptosis is a tightly controlled process of programmed cell death that plays an important role in many normal functions ranging from fetal development to adult tissue homeostasis [1]. Tumor is characterized by uncontrolled proliferation and reduced apoptosis. Activation of apopto- tic pathways is a key mechanism by which cytotoxic drugs kill cancer cells. Compounds that block or suppress the growth of tumor cells by inducing apoptosis are considered to have potential as anti-tumoral agents [2]. Herbal medicines derived from plant extracts are being increasingly used to treat a wide variety of clinical diseases. However, chemical constituents and definite mechanisms in many herbal medicines remained unknown, which hampered their further study and widespread agreement [3]. Actinidia valvata Dunn, whose root is known as ����mao ren shen�� in traditional Chinese medicine, exhibits anti-tumoral and anti-inflammatory activities and has been used for the treatment of hepatoma, lung carcinoma and myeloma for a long time [4,5]. Over the past 5 years, we carried out continuing studies mainly on its anti-tumoral pharmacology. A series of com- pounds have been found, among them were the triterpenoids [6]. The triterpenoids, synthesized in many plants by the cyclization of squalene, are widely used in Asian medicine [7]. Ursolic, pomolic, and maslinic acids were reported to have anti-tumoral properties [8�C11]. Here
Fig. 1), which is isolated from A. valvata Dunn and also has been discovered in many Chinese medicinal herbs, such as the Lagerstroemia speciosa L. [12] and banaba leaves [13]. It has been reported that CRA produces an excellent anti-diabetic activity in some animal experiments and clinical trials, including improvement of glucose metabolism by reducing insulin resistance in a mice model and lowering effect on post-challenge plasma glucose levels in human [12,14]. It was also reported that CRA displayed some cytotoxic activities against several human cancer cell lines [15,16]. However, the mechanisms for the anti-tumoral activity of CRA have not yet been explored. In this paper, we investigated the anti-cancer properties of CRA isolated fromA. valvata Dunn.We nowreport for the first time a study showing that CRA can induce apoptotic death in HeLa cells. This occurs through a mechanism that involves the deregulation of the cell cyclemachinery, direct depolarization of mitochondria, and activation of mitochondria-mediated, caspase-dependent apoptotic signaling cascades. Due to these effects, CRA may provide a useful new therapeutic strategy for cervix adenocarcinoma. 2. Materials and methods 2.1. Chemicals CRA was isolated from A. valvata Dunn. as described [6]. Briefly, powdered plant material of the roots of A. valvata Dunn. was refluxed in 80% ethanol. The extract was concentrated under reduced pressure and was partitioned between petroleum ether, ethyl acetate, n-butanol, and water successively. The ethyl acetate soluble fraction was subjected to column chromatography on silica gel, eluting with CHCl3/MeOH and was repeatedly subjected to CC (Pharmadex LH-20 and RP C-18) to yield CRA. CRA was prepared in sterilized dimethyl-sulfoxide (DMSO; Sigma) and stored at 4 C. Propidium iodide (PI) and 3-(4,5-dimethylthiazol-2-yl)- 2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma. The caspase fluorometric assay kits, z-VAD-FMK, MitoCaptureTM mitochondrial apoptosis detection kit and cytochrome c releasing assay kit were obtained from Biovision (Mountain View, CA). The annexin V-EGFP apoptosis detection assay kit and bicinchoninic acid assay kit were purchased from KeyGEN biotechnology (Nanjing,China) and Beyotime biotechnology (Haimen, China), respectively. Eagle��s Minimum Essential Medium and fetal calf serum (FCS) were obtained from GIBCO (Life Technologies, NY). Primary antibodies for b-actin, COX4, Bax and Bcl-2 were purchased from Cell Signaling (Danvers, MA). 2.2. Cell culture The human cervix adenocarcinoma cell line HeLa (ATCC No. CCL-2TM) was preserved by the Second Military Medical University and was cultured in Eagle��s Minimum Essential Medium supplemented with 10% FCS at 37 Cin5%CO2.In all experiments, cells were allowed to adhere and grow for 24 h in culture medium prior to exposure to CRA. During culture in the presence of CRA, we observed that an increasing proportion of cells were detached from the adhered monolayer and floated in culture supernatant. To determine the extent of apoptosis in our assays, we pooled both fractions, attached and floating cells. 2.3. Cell viability assay Cell viability was determined by measuring the absorbance of MTT dye staining of living cells, as described [17]. Briefly, cells were incubated in 100 ll of media in 96-well plates at an initial cell density of 5 [1] 104 cells/ ml. After 24 h incubation, medium was removed and replaced by 10% FCS medium containing indicated CRA. An appropriate volume of drug vehicle was added to untreated cells. After a period time of incubation, 10 ll MTT solution (5 mg/ml in PBS) was added and cells were incubated for a further 4 h. One hundred microliters of SDS�Cisobutanol�CHCl solution (10% SDS, 5% isobutanol, and 12 lM HCl) were added to each culture and incubated overnight. Relative cell viability was obtained by scanning with a microplate reader (Bio-Rad, San Diego, CA) with a 595 nm filter. Five wells per dose were counted in three independent experiments. 2.4. Cell-cycle analysis Cells were seeded at 2 [1] 105 in six-well plates, cultured in 10% FCS medium with or without CRA. After the treatment, adherent and floating cell populations were combined and fixed in ethanol (70% in PBS), washed in PBS and stained with PI (20 lg/ml in final concentration). Flow cytometric analyses were performed using a FACSCalibur cytometry (Becton Dickinson, San Jose, CA). Cell distribution in the different phases of the cell cycle was analyzed with ModFit LT V3.0 software. 2.5. Analysis of apoptotic cells After exposure to CRA, the apoptotic cell death was determined using an annexin V-EGFP apoptosis detection assay kit according to the modified manufacturer��s protocol. Cells were seeded on 12-mm glass cover-slips Fig. 1. Structure of corosolic acid (2a,3b-dihydroxy-urs-12-en-28-oic acid). 230 Y. Xu et al. / Cancer Letters 284 (2009) 229�C237at a density of 1 [1] 105 cells/ml and were treated with 40 lM of CRA for 24 h. After being incubated, the coverslips were washed with PBS, 100 ll of binding buffer containing 2 ll annexin V-EGFP was added to each cover-slips and then incubated at room temperature for 15 min in dark. PI in a final concentration of 20 lg/ml was added and incubated for another 5 min. After being washed with PBS, the cover-slips were visualized under fluorescence microscopy (Olympus, Tokyo, Japan). The CRA-induced apoptotic effect was also determined with flow cytometry. Briefly, after the incubation, adherent and floating cell populations were combined, rinsed with PBS, and resuspended in 200 ll of binding buffer at a concentration of 1 [1] 106 cells/ml. Five microliters of annexin V-EGFP were added and incubated for 15 min in dark. Then PI was added and an immediate analysis was performed with a FACSCal- ibur cytometry. 2.6. Detection of caspase catalytic activities The activities of caspase-8, -9, -12, and -3 were studied using the caspase fluorometric assay kits. Assays are based on fluorometric measurement of fluorescent 7-amino-4-trifluoromethyl coumarin (AFC) after cleavage from the AFC-labeled peptide substrates, IETD-AFC for caspase-8, LEHD-AFC for caspase-9, ATAD-AFC for caspase-12 and DEVD-AFC for caspase-3 [18]. Briefly, after being treated with 40 lM of CRA for the indicated time, cells were harvested and kept on ice. Cell lysis buffer was added to the cell pellets, and the protein concentration was determined using a bicinchoninic acid assay [19]. Fifty micrograms of each cell lysate was incubated with 2 ll of 1 mM stock of fluorescently labeled tetrapeptide caspase substrate at 37 C for 30 min. The release of AFC was measured with a fluorometric plate reader (Molecular devices, Sunnyvale, CA) at an excitation wavelength of 400 nmand an emission wavelength of 505 nm. Experiments were performed in triplicate. 2.7. Detection of mitochondrial transmembrane potential disruption MitoCapture mitochondrial apoptosis detection kit was used for detection of Dwm disruption. MitoCapture accumulates and aggregates in normal mitochondrial, giving off a bright red fluorescence. In apoptotic cells, MitoCapture can not aggregate in the mitochondria due to the altered Dwm, and thus it remains in the cytoplasm in its monomer form, fluorescing green. Cells were seeded on 12-mm glass cover-slips at a density of 1 [1] 105 cells/ml and were treated with CRA or medium with vehicle for controls. After incubation with CRA, 100 ll pre-warmed incubation buffer containing 0.1 llMitoCapture was added to each cover-slips and incubated for 15 min at 37 C. After being washed with PBS, the cover-slips were mounted and the fluorescence was visualized under a fluorescent microscopy. The disruption of Dwm was also quantified by flow cytometry. After treated with CRA, 1 [1] 106 cells were collected and resuspended in 1 ml pre-warmed incubation buffer containing 1 ll MitoCapture. After 15 min at 37 C incubation, the cells were resuspended in incubation buffer and analyzed immediately by flow cytometry in the FITC channel. At least 1 [1] 104 cells were analyzed for each sample. 2.8. Protein extraction and Western blot analysis For detection the releasing of cytochrome c from mitochondria into cytosol, a cytochrome c releasing assay kitwas used. The cytosolic and mitochondrial fraction proteins were collected according to the manufacturer direction. For Bax and Bcl-2 immunoblot analysis, protein extracts were obtained by washing cells with PBS and suspending in RIPA lysis and extraction buffer containing protease inhibitor cocktail (Pierce, Rockford, IL). Immunoblot assays were performed as described [20]. Briefly, equal amounts of protein as determined by a bicinchoninic acid assay were subjected to SDS�CPAGE, followed by transfer to PVDF membranes (Millipore, Bedford, MA). Membranes were then blocked in 5% powdered non-fat milk in a Tris/ Tween solution (20 mM Tris�CHCl, pH 7.6, 0.1% Tween 20 in saline) for 2 h. The primary antibodies were diluted
Fig. 2. Effect of CRA on the viability of HeLa cell. (A) Cells were treated with various concentrations of CRA and relative cell viability was assessed by MTT assay for 24 h (diamond), 48 h (square) and 72 h (triangle). Results were expressed as percentage of untreated controls �� SD obtained from three separated experiments. (B) Growth inhibition and morphologic changes of HeLa cells treated with 40 lM for 12 h, 24 h or 36 h, compared with untreated cells (control). Cells were photographed with a Leica DM IL inverted contrasting microscopy (100[1]). Y. Xu et al. / Cancer Letters 284 (2009) 229�C237 231(1:200 for cytochrome c; 1:1,000 for b-actin, COX4, Bax and Bcl-2) in blocking solution and then incubated with the membrane over night at 4 C. The membranes were then covered with an HRP conjugated secondary IgG antibody (goat anti-mouse for cytochrome c, goat anti-rabbit for b-actin, Bax, Bcl-2 and COX4) at a 1:2,500 dilution for 2 h. Detection was performed by an enhanced chemilumi-nescence method (Pierce, Rockford, IL). 2.9. Statistical analysis Data are expressed as the means �� SD. For each assay, Student��s t-test was used for statistical comparison. A probability of p < 0.05 was considered significant. 3. Results 3.1. CRA decreased cell viability of HeLa We examined the effect of CRA on cell viability using MTT assay. HeLa cells were treated with various concentrations of CRA, their viability was determined by formazan dye uptake and expressed as percent of untreated control cells. CRA induced both a dose- and a time-dependent decrease in viable formazan accumulating cells after the treatment (Fig. 2A). The 50% growth inhibition concentration (IC50), obtained after 24 h, 48 h and 72 h of incubation were 45 lM, 34 lM and 28 lM, respectively. In addition, cell morphology was also examined using a phase-contrast microscope. Microscopic observations revealed that CRA affected not only cell growth but also cell appearance and attachment (Fig. 2B). Exposure of HeLa cells to 40 lM CRA provoked significant morphology alterations. For 6 h, HeLa cells began to shrink and retract from their neighbors (data not shown). For 12 h, the floating cells appeared in the culture medium. At 36 h of incubation with CRA, most of the HeLa cells lost their flat, polygonal shape. Meanwhile, the number of survival cells decreased significantly when compared with the control. 3.2. CRA induced cell cycle block The cytotoxicity caused by CRA may be due in part to anti-proliferative and proapoptotic effects. The effect of CRA on cell cycle progression was analyzed by flow cytometry. HeLa cells were treated with CRA and their distribution in the different phases of the cell cycle was calculated (Fig. 3). At 24 h, cells in the S phase of the cell cycle increased from 35.64% to 48.01% in a dose-dependent manner (Fig. 3A). When treated with 40 lM of CRA, the S phase cells increased from 35.28% to 46.57% in a time-dependent manner (Fig. 3B). A slight decrease in the population of cells in G0/G1 phase can also be figured out. All these findings indicated that CRA caused a late S phase or early G2/M block. The block in cell cycle progression, therefore, contributes to CRA induced anti-proliferative ef- fects. In addition, there was a significant increase in the sub-G0 fraction (hypodiploid DNA content), possibly due to DNA fragmentation, resulting in an increase in CRA-induced apoptotic cell death (Fig. 3C and D). 3.3. CRA-induced apoptosis of HeLa Since a time- and a dose-dependent sub-G0 fraction appeared in the cell-cycle analysis, CRA-induced apoptosis was further confirmed using annexin V-EGFP/PI double staining. Treated with 40 lM of CRA for 24 h, the apoptotic cells membrane was stained by green color (annexin V+)and chromatin fragmentation stained by red color (PI+). In contrast, the control cells were stained neither by annexin V nor PI (Fig. 4A). CRA-in-duced apoptosis of HeLa cells was also determined by flow cytometry(Fig. 4B). Fig. 3. Cell-cycle analysis. HeLa cells were cultured with CRA and the cell phase distribution was determined by PI staining and analyzed using ModFit LT software. Results were obtained from three separated experiments. (A) The S phase cells increased from 35.64% in the control to 37.09%, 38.81% and 48.01% when treated with various concentration of CRA for 24 h. (B) The S phase cells increased from 35.28% in the control to 38.84%, 39.72% and 46.57% when treated with 40 lM of CRA for 6 h, 12 h and 18 h. (C) The sub-G0 fraction increased from 0.46% in the control to 15.05%, 21.66% and 25.84% when treated with various concentration of CRA for 24 h. (D) The sub-G0 fraction increased from 0.24% in the control to 5.18%, 10.29% and 18.02% when treated with 40 lM of CRA for 6 h, 12 h and 18 h. Significant difference from control value was indicated (p < 0.01). 3.4. CRA treatment induced caspases activation The involvement of caspase pathway in apoptosis has been established in many studies. To examine whether the apoptotic effects induced by CRA were associated with caspase enzymes activation or not, the activities of initiator caspase (caspase-8, -9, and -12) and effector caspase (cas-pase-3) were investigated by fluorometric protease assay. HeLa cells were left untreated or treated with 40 lM of CRA, and caspase activities were determined. The fluorometric protease assay showed a time-dependent increase in the activities of caspase-8, -9, and -3 in CRA-treated cell ly- sates, while the activity of caspase-12 remained unchanged (Fig. 5). Slight increases in caspase-8, -9 and -3 activities were observed 6 h after the addition of CRA. The activities of caspase-8 and caspase-9 increased 1.6-fold and 2.3-fold after 12 h, 2.3-fold and 3.4-fold after 24 h treatment, respectively (Fig. 5A and B). Caspase-3 activity increased 9.6-fold after 12 h and 14.9-fold after 24 h treatment (Fig. 5D). The slight increase in caspase-12 activity at 24 h did not reach statistical significance(Fig. 5C). These results indicated that caspase-8, -9, and -3, but not caspase-12, were involved in the apoptotic effects induced by CRA. Treatment of cells with 10 lM of z-VAD-FMK, a general inhibitor of caspases, 1 h before CRA added markedly attenuated CRA-induced apop- tosis. As shown in Fig. 5E, apoptosis was observed in about 60.7% at 24 h following CRA treatment in the absence of z-VAD-FMK, while it was observed in only 10.6% in the presence of z-VAD-FMK. These results clearly indicated that CRA-induced apoptosis was dependent on caspase activation. 3.5. CRA treatment induced alterations in Dwm An observation on variations for caspase-8 and caspase-9 prompted us to study the drop in Dwm. The MitoCapture kit was used for detection of alterations in Dwm on viable adherent cells. At 12 h, the control cells gave off a bright red fluorescence when using a rhodamine filter. While in the CRA-treated cells, the majority of cytoplasm fluoresced green when a FITC filter was used (Fig. 6A). Then we analyzed the Dwm in CRA treated HeLa cells using flow cytometry. CRA treatment caused the loss of Dwm in a time-dependent manner (Fig. 6B), as shown by the shift in the cell population from low to high green fluorescence. 3.6. CRA treatment induced cytochrome c release from the mitochondria Mitochondria play an essential role in apoptosis triggered by chemical agents. The mitochondrial response includes the release of cytochrome cinto the cytosol. In the cytosol, cytochrome c binds to Apaf-1, allowing the recruitment of caspase-9 and the formation of an apoptosome complex, resulting in caspase-3 activation and execution of cell death [21]. To analyze the involvement of mitochondrial release of cytochrome c in HeLa cells, proteins from both cytosolic and mitochondrial fractions were prepared and analyzed with western blot.We introduced COX4 for the mitochondrial fractions and b-actin for the cytosolic fraction as internal controls (Fig. 6C). Treatment of HeLa cells with 40 lM of CRA caused a gradual decrease in mitochondrial cytochrome c, with a concomitant increase in the cytosolic fraction. The release of cytochrome c was detected as early as 3 h post-treatment. However, the level of COX4 and b-actin remained unchanged. These results indicate that CRA can provoke cytochrome c release from mitochondria into cytosol. 3.7. CRA-induced apoptosis with an increase in Bax/Bcl-2 ratios As we demonstrated a time-dependent drop in Dwm and release of cytochrome c from mitochondria in HeLa cells treated with CRA, it was interesting to study the proapoptotic and anti-apoptotic members linked to mitochondrial control during apoptosis: Bax and Bcl-2. The expressions of Bax and Bcl-2 protein were obtained by western blot analysis. The expression of b-actin was monitored to ensure that equal amounts of protein samples were loaded in all lanes. As seen in Fig. 6D, CRA treatment of HeLa cells resulted in an increased level of Bax expression in a time-dependent manner. This expression was detected as early as 3 h post CRA treatment. The level of the anti-apoptotic protein Bcl-2 remained unchanged during the CRA exposure time. And CRA did not affect the level of b-actin protein, used as a loading and an internal control. These results suggest that CRA-induced apoptosis in HeLa cells via alteration of the Bax/Bcl-2 ratio. 4. Discussion During the past decades, much effort has been made toward the search for compounds or herbs that kill tumors through induction of apoptosis [2,22]. Triterpenoids exerts various pharmacological effects to control many diseases, including cancer [10,16,23�C25]. The present study is the first to evaluate the potential anti-tumoural effects of CRA, a new triterpenoid isolated as a majority compound from A. valvata Dunn. Our data showed that CRA exhibited anti-proliferative and proapoptotic effects on HeLa cervix adenocarcinoma cell line in both a time- and a dose-dependent manner. By MTT assay, we demonstrated that CRA significantly suppressed HeLa cell viability. It could also induce mor- phological changes that were characteristic of apoptosis, such as cell shrinkage and detachment, which were observed under a phase-contrast microscopy. Stained with PI and analyzed by flow cytometry, our results indicated that CRA inhibited HeLa cell proliferation by inducing late S phase or early G2/M growth arrest. The appearance of a Y. Xu et al. / Cancer Letters 284 (2009) 229�C237 233sub-G0 cell population indicated that CRA-induced apoptosis in HeLa cells. However, the role of cell cycle regulation on the extent of CRA-induced apoptosis requires further demonstration. Double stained with annexin V/PI, and observed under a fluorescent microscopy, CRA was found to induce morphological changes characteristic of apoptosis, such as the translocation of phosphatidylserine from the inner to the outer membrane and loss of cell-membrane integrality. The percentage of apoptosis quantified by flow cytometry analysis showed that 28.4% of the HeLa cells showed signs of cell apoptosis after 12 h treatment and 60.7% after 24 h. Because the execution of apoptosis appears to be uniformly mediated through consecutive activation of the members of a caspase family [26], we examined whether the possible apoptotic effect of CRA on HeLa cells could be linked to induction of caspase activation. Our data showed that as early as 6 h treatment, considerable caspase-8, -9 and -3 activities were observed, but not cas-pase-12. Moreover, CRA-induced apoptosis was dependent on caspase activation since apoptosis was mostly inhibited by the general caspase inhibitor z-VAD-FMK. Caspase-12 was proved to play a role in endocytoplasmic reticulum stress-mediated cell death [27]. Our results indicated that endocytoplasmic reticulum may not involved in CRA-induced apoptosis in HeLa cells. Caspase-dependent process is associated with two pathways, i.e., a death receptor-dependent and a mitochondria-dependent path-way. Death-receptor activation pathway is mediated with a death-inducing signaling complex, which is made of a Fas-associated death domain and a procaspase-8, activating caspase-8 and leading to the activation of the down-stream caspases [28]. The mitochondria-dependent pathway for apoptosis involves the release of cytochrome
Fig. 5. CRA-induced apoptosis is mediated by caspase activation. (A) Caspase-8 activity; (B) Caspase-9 activity; (C) Caspase-12 activity; (D) Caspase-3 activity. The results were measured spectrofluorometrically using AFC-labeled peptide as substrates. The lysates were adjusted to equal protein concentrations using the bicinchoninic acid assay. (E) z-VAD-FMK inhibited CRA-induced apoptosis. Cells pre-treated with 10 lM z-VAD-FMK were incubated with 40 lM of CRA for 24 h, double stained with annexin V/PI and was measured by flow cytometry. Values are expressed as means �� SD of triplicate samples. Results are representative of one of three independent experiments. Significant difference from control value was indicated by (p < 0.05) Or (p < 0.01). Significant difference from CRA treatment value was indicated by ##(p < 0.01). 234 Y. Xu et al. / Cancer Letters 284 (2009) 229�C237c from mitochondria into the cytosol [29], where it binds to apoptotic protease activating factor 1 (Apaf-1), allowing the recruitment of caspase-9 and the formation of an apoptosome complex, resulting in caspase-3 activation and execution of cell death [21]. In addition, the activation of caspase-8 can directly cleave Bid, a proapoptotic member of the Bcl-2 family, to trigger cytochrome c release [30]. To confirm whether CRA-induced apoptosis through a mitochondria-dependent pathway, we analyzed the disruption of Dwm and cytochrome c levels both in mitochondria and in cytosol. In situ labeling of mitochondria by Mitocapture showed that CRA induced a drop in Dwm in HeLa cells. Flow cyto- metric results confirmed this with mitocapture in situ labeling and indicated that CRA induced an early drop in Dwm at 6 h. By western blot, we found that cytochrome c showed a gradual decrease in mitochondria, with a concomitant increase in cytosol. And the release of cytochrome c was detected as early as 3 h post-treatment, which precedes the activation of caspases. The release of cytochrome c and cytochrome c mediated apoptosis is controlled by multiple layers of regulation, the most prominent players being members of Bcl-2 family. Bax and Bcl-2 have been identified as major regulators in controlling the release of mitochondrial cytochrome c. Bax translocates to the mitochondria, and inserts into the outer mitochondrial membrane, where allows the release of cytochrome c. In contrast, Bcl-2 blocks cytochrome c efflux by binding to the outer mitochondrial membrane [31]. Many anti-cancer agents can trigger the release of cytochrome c through either up-regulation of Bax and/or down-regulation of Bcl-2 [10,20,32]. After HeLa cells were treated with CRA, Bax expression significantly increased, while Bcl-2 expressionmaintained. This result showed that balance between Bax and Bcl-2 was upset in favor of Bax. Hence, increasing Bax vs. Bcl-2 suggests the mitochondrial permeability transition pore opening and induction of apoptosis. In conclusion, CRA showed a significant anti-proliferative effect on HeLa cells. To our knowledge, these results clearly showed for the first time that CRA induces apoptosis in cervix adenocarcinoma cells. CRA triggered the apoptotic pathway characterized by Bax overexpression, collapse of mitochondrial membrane potential, and subsequent release of cytochrome c from mitochondria into cytoplasm. This apoptotic pathway is also characterized by the caspase cascade (via caspase-8 and -9) which culminated in caspase-3 activation (Fig. 7). Triterpenoids such as ursolic acid, pomolic acid, and maslinic acid have been reported to induce apoptosis by similar mechanisms [8�C11].
Fig. 6. CRA induced HeLa cells apoptosis through a mitochondrial pathway. (A) HeLa cells were seeded on cover-slips and treated with 40 lM of CRA for 12 h, then stained with Mitocapture and photographed with a Olympus fluorescent microscopy (400[1]). (B) HeLa cells were induced with 40 lM of CRA for 6 h or 12 h, then incubated with Mitocapture for 15 min. Results were analyzed by flow cytometry using FITC channel. (C) CRA treatment induced cytochrome c release from the mitochondria. HeLa cells were treated with 40 lM of CRA for the indicated time periods. The cytosolic and mitochondrial fraction proteins were collected using a cytochrome c releasing assay kit. Cytochrome c releasing from mitochondria into cytosol was then determined by western blot using the cytochrome c antibody provided in the kit. COX4 and b-actin were used as internal controls for the mitochondrial fractions and the cytosolic fraction, respectively. (D) CRA treatment modulates Bax and Bcl-2 protein expression in HeLa cells. HeLa cells were treated with 40 lM of CRA for the indicated time periods. Cell extracts were made, resolved by SDS�CPAGE and immunoanalyzed for Bax, Bcl-2 or b-actin with corresponding protein-specific antibodies. Y. Xu et al. / Cancer Letters 284 (2009) 229�C237 235
Fig. 7. Mechanisms of CRA-induced apoptosis. CRA up-regulates Baxexpression and activates caspase-8, resulting in mitochondrial depolarization that is frequently associated with mPTP opening. Subsequent mitochondrial release of cytochrome c leads to activation of caspase-9 and -3, resulting in apoptosis. Caspase-8 itself can activation of caspase-3, resulting in apoptosis. 236 Y. Xu et al. / Cancer Letters 284 (2009) 229�C237 Maslinic acid, which is an isomer of CRA, induced reactive oxygen species generation and subsequently leading astrocytoma cell apoptosis [11]. Considerable evidence also indicates that reactive oxygen species can enhance spontaneous apoptosis in which caspase-8 is involved in the signaling pathway [33,34]. Whether CRA triggers cas-pase-8 activation is associated with oxidative stress and the precise signaling pathway by which CRA up-regulates Bax expression remained to be identified. Nevertheless, we hope that our findings could contribute to the develop- ment of corosolic acid and related drugs for use as cancer chemotherapeutic or chemopreventive agents. 5. Conflicts of interest None declared. Acknowledgements This work was supported by the National Natural Science Foundation of China (Grant No. 30572360) and Special Project of TCM Modernization of Science and Technology Commission of Shanghai Municipality (Grant No. 04DZ19808). We thank Prof. Mingyong Miao and Prof. Ying Hou for helping in preparation of the manuscript. Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.canlet.2009. 04.028. References [1] J.C. Reed, Apoptosis-regulating proteins as targets for drug discovery, Trends Mol. Med. 7 (2001) 314�C319. [2] O.S. Frankfurt, A. Krishan, Apoptosis-based drug screening and detection of selective toxicity to cancer cells, Anticancer Drugs 14 (2003) 555�C561. [3] M.C. Wu, Traditional Chinese medicine in prevention and treatment of liver cancer: function, status and existed problems, J. Chin. Integr. Med. 1 (2003) 163�C164. [4] Z.Z.Wang, Y. Song, J.H. Hu, B.B. You,Morphological identification and the clinical application of the roots of Actinidia chinensis and Actinidia valvata, Pharm. Care Res. 5 (2005) 134�C137. [5] Y.N. Zhang, L. Liu, C.Q. Ling, Inhibition effect of active fraction from Actinidia valvata on growth of transplanated mouse tumor cells and preliminary study of its mechanism, Zhongguo Zhong Yao Za Zhi 31 (2006) 918�C920. [6] H.L. Xin, X.Q. Yue, Y.F. Xu, Y.C. Wu, Y.N. Zhang, Y.Z. Wang, C.Q. Ling, Two new polyoxygenated triterpenoids from Actinidia valvata, Helv. Chim. Acta 91 (2008) 575�C580. [7] K.T. Liby, M.M. Yore, M.B. Sporn, Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer, Nat. Rev. Cancer 7 (2007) 357�C369. [8] P.O. Harmand, R. Duval, C. Delage, A. Simon, Ursolic acid induces apoptosis through mitochondrial intrinsic pathway and caspase-3 activation in M4Beu melanoma cells, Int. J. Cancer 114 (2005) 1�C11. [9] J. Fernandes, R. Weinlich, R.O. Castilho, M.A. Kaplan, G.P. Amarante- Mendes, C.R. Gattass, Pomolic acid triggers mitochondria-dependent apoptotic cell death in leukemia cell line, Cancer Lett. 219 (2005) 49�C55. [10] F.J. Reyes-Zurita, E.E. Rufino-Palomares, J.A. Lupianez, M. Cascante, Maslinic acid, a natural triterpene from Olea europaea L., induces apoptosis in HT29 human colon-cancer cells via the mitochondrial apoptotic pathway, Cancer Lett. 273 (2009) 44�C54. [11] R. Martin, J. Carvalho, E. Ibeas, M. Hernandez, V. Ruiz-Gutierrez, M.L. Nieto, Acidic triterpenes compromise growth and survival of astrocytoma cell lines by regulating reactive oxygen species accumulation, Cancer Res. 67 (2007) 3741�C3751. [12] M. Fukushima, F. Matsuyama, N. Ueda, K. Egawa, J. Takemoto, Y. Kajimoto, N. Yonaha, T. Miura, T. Kaneko, Y. Nishi, R. Mitsui, Y. Fujita, Y. Yamada, Y. Seino, Effect of corosolic acid on postchallenge plasma glucose levels, Diabetes Res. Clin. Pract. 73 (2006) 174�C177. [13] Y. Yamaguchi, K. Yamada, N. Yoshikawa, K. Nakamura, J. Haginaka, M. Kunitomo, Corosolic acid prevents oxidative stress, inflammation and hypertension in SHR/NDmcr-cp rats, a model of metabolic syndrome, Life Sci. 79 (2006) 2474�C2479. [14] T. Miura, N. Ueda, K. Yamada, M. Fukushima, T. Ishida, T. Kaneko, F. Matsuyama, Y. Seino, Antidiabetic effects of corosolic acid in KK-Ay diabetic mice, Biol. Pharm. Bull. 29 (2006) 585�C587. [15] K.S. Ahn, M.S. Hahm, E.J. Park, H.K. Lee, I.H. Kim, Corosolic acid isolated from the fruit of Crataegus pinnatifida var. psilosa is a protein kinase C inhibitor as well as a cytotoxic agent, Planta Med. 64 (1998) 468�C470. [16] M. Yoshida, M. Fuchigami, T. Nagao, H. Okabe, K. Matsunaga, J. Takata, Y. Karube, R. Tsuchihashi, J. Kinjo, K. Mihashi, T. Fujioka, Antiproliferative constituents from Umbelliferae plants VII. Active triterpenes and rosmarinic acid from Centella asiatica, Biol. Pharm. Bull. 28 (2005) 173�C175. [17] J.J. Zhou, X.F. Yue, J.X. Han, W.Y. Yang, Improved MTT assay for activity of antitumor agents, Chin. J. Pharmaceut. 24 (1993) 455�C 457. [18] B.S. Cummings, R.G. Schnellmann, Cisplatin-induced renal cell apoptosis: caspase 3-dependent and -independent pathways, J. Pharmacol. Exp. Ther. 302 (2002) 8�C17. [19] M.J. Liu, P.Y. Yue, Z. Wang, R.N. Wong, Methyl protodioscin induces G2/M arrest and apoptosis in K562 cells with the hyperpolarization of mitochondria, Cancer Lett. 224 (2005) 229�C241. [20] A. Das, N.L. Banik, S.K. Ray, Mechanism of apoptosis with the involvement of calpain and caspase cascades in human malignant neuroblastoma SH-SY5Y cells exposed to flavonoids, Int. J. Cancer 119 (2006) 2575�C2585. [21] D.R. Green, J.C. Reed, Mitochondria and apoptosis, Science 281 (1998) 1309�C1312. [22] L.S. Einbond, Y. Wen-Cai, K. He, H.A. Wu, E. Cruz, M. Roller, F. Kronenberg, Growth inhibitory activity of extracts and compounds from Cimicifuga species on human breast cancer cells, Phytomedicine 15 (2008) 504�C511. [23] A.K. Pathak, M. Bhutani, A.S. Nair, K.S. Ahn, A. Chakraborty, H. Kadara, S. Guha, G. Sethi, B.B. Aggarwal, Ursolic acid inhibits STAT3 activation pathway leading to suppression of proliferation and chemosensitization of human multiple myeloma cells, Mol. Cancer Res. 5 (2007) 943�C955. [24] S.Y.Wei,M. Li, S.A. Tang,W. Sun, B. Xu, J.R. Cui,W.H. Lin, Induction of apoptosis accompanying with G(1) phase arrest and microtubule disassembly in human hepatoma cells by jaspolide B, a new isomalabaricane-type triterpene, Cancer Lett. 262 (2008) 114�C122. [25] A.M.H.O. Yasutaka Ikeda, Ursolic acid: an anti- and proinflammatory triterpenoid, Mol. Nutr. Food Res. 52 (2008) 26�C42. [26] A. Philchenkov, Caspases: potential targets for regulating cell death,J. Cell Mol. Med. 8 (2004) 432�C444. [27] T. Nakagawa, H. Zhu, N. Morishima, E. Li, J. Xu, B.A. Yankner, J. Yuan,Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta, Nature 403 (2000) 98�C103. [28] S.G. Cho, E.J. Choi, Apoptotic signaling pathways: caspases and stress-activated protein kinases, J. Biochem. Mol. Biol. 35 (2002) 24�C27. [29] V. Gogvadze, S. Orrenius, B. Zhivotovsky, Multiple pathways of cytochrome c release from mitochondria in apoptosis, Biochim. Biophys. Acta 1757 (2006) 639�C647. [30] D.R. Green, Apoptotic pathways: the roads to ruin, Cell 94 (1998) 695�C698. [31] Y.P. Ow, D.R. Green, Z. Hao, T.W. Mak, Cytochrome c: functions beyond respiration, Nat. Rev. Mol. Cell Biol. 9 (2008) 532�C542. [32] M.A. El-Mahdy, Q. Zhu, Q.E. Wang, G. Wani, A.A. Wani, Thymoquinone induces apoptosis through activation of caspase-8 and mitochondrial events in p53-null myeloblastic leukemia HL-60 cells, Int. J. Cancer 117 (2005) 409�C417. [33] D. Scheel-Toellner, K. Wang, R. Craddock, P.R. Webb, H.M. McGettrick, L.K. Assi, N. Parkes, L.E. Clough, E. Gulbins, M. Salmon, J.M. Lord, Reactive oxygen species limit neutrophil life span by activating death receptor signaling, Blood 104 (2004) 2557�C2564. [34] W. Zhang, I. Hirschler-Laszkiewicz, Q. Tong, K. Conrad, S.C. Sun, L. Penn, D.L. Barber, R. Stahl, D.J. Carey, J.Y. Cheung, B.A. Miller, TRPM2 is an ion channel that modulates hematopoietic cell death through activation of caspases and PARP cleavage, Am. J. Physiol. Cell Physiol. 290 (2006) C1146�C1159. Y. Xu et al. / Cancer Letters 284 (2009) 229�C237 237 |



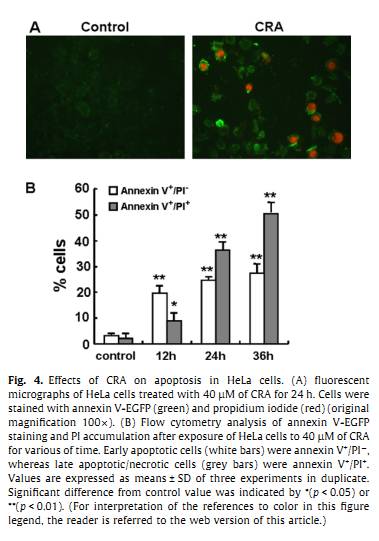 In comparisonwith untreated controls, HeLa cells generated apoptosis in 28.4% of cells when treated with 40 lM of CRA for 12 h (19.5% early apoptosis plus 8.9% late apoptosis/necrosis), in 60.7% of cells for 24 h (24.5% early apoptosis plus 36.2% late apoptosis/necrosis) and in 77.8% of cells for 36 h (27.4% early apoptosis plus 50.4% late apoptosis/necrosis). Thus the annexin V/PI double staining assay confirmed the observation that CRA-in-duced apoptosis.
In comparisonwith untreated controls, HeLa cells generated apoptosis in 28.4% of cells when treated with 40 lM of CRA for 12 h (19.5% early apoptosis plus 8.9% late apoptosis/necrosis), in 60.7% of cells for 24 h (24.5% early apoptosis plus 36.2% late apoptosis/necrosis) and in 77.8% of cells for 36 h (27.4% early apoptosis plus 50.4% late apoptosis/necrosis). Thus the annexin V/PI double staining assay confirmed the observation that CRA-in-duced apoptosis.

